 Hotline服務熱線:010-61006450
Hotline服務熱線:010-61006450
 簡體中文
簡體中文文獻解讀 | 用生理學模型評價食物對克拉霉素速釋片口服吸收的影響
生理藥代動力學(PBPK)等模型的建立有助于預測體外特征對藥物吸收的影響。本文以《Evaluation of food effect on the oral absorption of clarithromycin from immediate release tablet using physiological modelling》文獻為依據,通過生理吸收模型預測克拉霉素速釋片在體內吸收的曲線及分布,同時采用機械吸收模型建立體內外相關性(IVIVR),詳細解讀pH、膽汁分泌和食物對克拉霉素速釋片口服吸收的影響。
體外檢測克拉霉素在各介質中的溶解度、片劑崩解和溶出曲線,以及高脂餐勻漿后的流變。
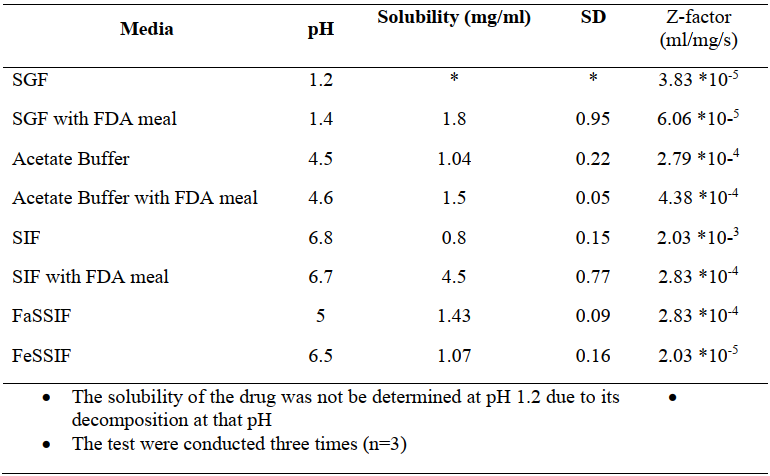
? 降解:克拉霉素在pH3-8水溶液中較為穩定,在胃pH(1-2)下會迅速降解。室溫下,在0.1N HCl介質中1h內便會降解50%。
? 溶解度:克拉霉素的溶解度無pH依賴性,由于克拉霉素在酸性條件下會快速降解,無法準確測得pH1.2的溶解度。檢測數據表明,由于高脂餐中的高脂肪含量可顯著提高親脂性藥物(BCSII類)的溶解度,食物會促進克拉霉素的溶解。
表1 克拉霉素在不同介質中的溶解度和轉換因子
? 流變:稀釋后高脂餐的流變性結果表明,食物的流變性具有假塑性,隨剪切速率增加粘度降低。高脂餐的粘度具有pH依賴性,粘度與pH呈正相關。同時,在SIF和醋酸鹽介質中,高脂餐的流變曲線具有疊加性。
? 崩解:介質pH值對克拉霉素片的崩解影響顯著,由于片劑表面會生成凝膠,克拉霉素片在酸性介質中崩解時間較堿性介質中偏長。在不同介質中加入食物,也會延遲藥物的崩解,這是由于加入食物后介質粘度變高。
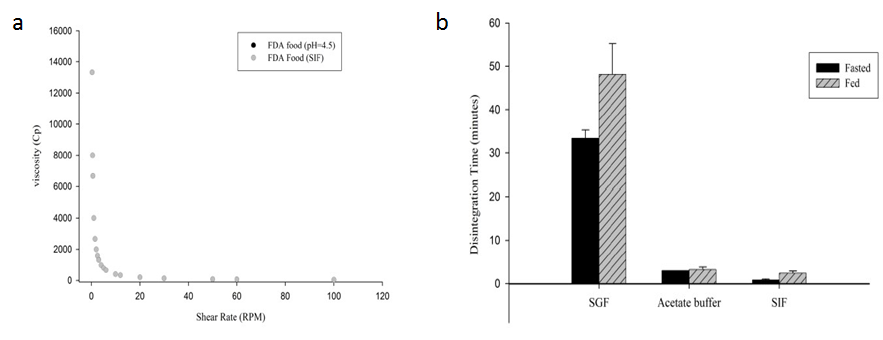
圖1 a:標準高脂餐(醋酸鹽和磷酸鹽緩沖液)在不同剪切速率下的粘度;b:克拉霉素在不同介質中的平均崩解時間
? 體外藥物釋放影響因素:
-
介質pH:克拉霉素片在pH4.5和pH6.8介質中溶出較為完全,但在pH1.2介質中溶出終點僅為15%。這是因為克拉霉素在酸性介質中發生降解,且片劑表面形成凝膠而減慢了克拉霉素片的崩解。
-
食物:食物對藥物的體外釋放影響顯著,但各pH中食物對藥物釋放的影響不盡相同。在人工胃液(SGF)中食物會促進克拉霉素釋放,減緩降解速率。然而,在pH4.5和pH6.8介質中,片劑崩解緩慢,食物降低了克拉霉素的吸收。
-
膽酸鹽:測定克拉霉素在生物相關介質和常規介質中釋放。克拉霉素在禁食狀態腸液和禁食狀態胃液介質中釋放均比常規介質中偏低。在禁食狀態腸液(pH6.5)和磷酸鹽緩沖液(pH6.8)介質中120min分別釋放60%、88%,兩個介質pH值僅差0.3,但釋放百分比相差28%。克拉霉素在禁食狀態胃液(pH=5)釋放非常低,120min僅釋放10%。對比空腹和餐后狀態各介質中的藥物釋放,f1和f2均小于50,二者不等效。
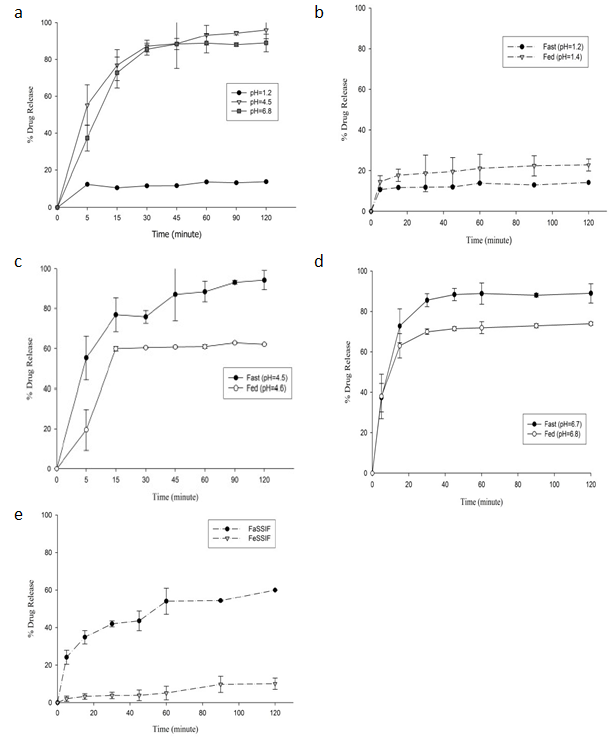
圖2 a:克拉霉素速釋片(500 mg)在各介質中的溶出曲線;b:克拉霉素在胃液(空腹和餐后)介質中溶出曲線;c:克拉霉素在醋酸鹽(空腹和餐后)介質中溶出曲線;d:克拉霉素在磷酸鹽緩沖液(空腹和餐后)介質中溶出曲線;e:克拉霉素在生物相關介質中的體外溶出曲線(禁食狀態腸液與禁食狀態胃液)
01、藥物吸收建模:
使用GastroPlus軟件PBPK模型對口服給藥的克拉霉素500mg速釋片進行建模,部分建模參數如表2,其他參數為軟件默認。模型預測參數與觀測值詳見表3,經比較,預測值與觀測值誤差不超過10%,表明此次建模具有較好的預測性。由于食物減慢了胃排空速率,餐后給藥Tmax延長。同時,模擬結果表明食物對Cmax和AUC無顯著性影響。
表2 建模輸入參數
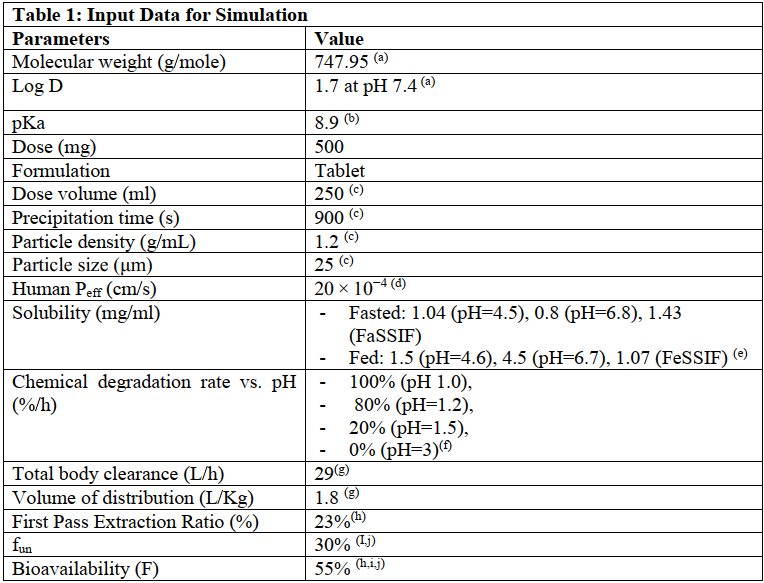
表3 PK參數的建模預測值與觀測值
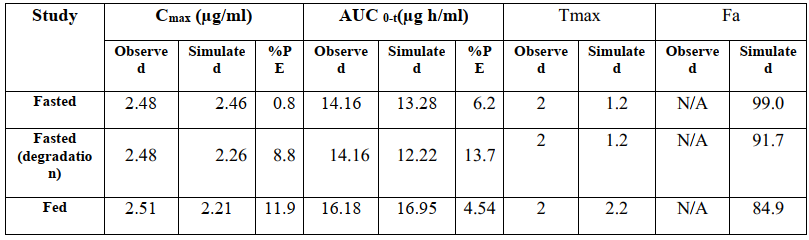
研究藥物的酸降解對克拉霉素口服生物利用度的影響發現,考慮酸降解時Cmax和AUC減小。克拉霉素在胃腸道吸收快速,且模型預測空腹條件下克拉霉素吸收分數為91.6%,餐后條件下吸收100%。考慮原因為克拉霉素在胃pH環境中不穩定,降低了藥物的吸收。空腹狀態下克拉霉素67.1%吸收分布在十二指腸,27.1%的劑量在空腸。餐后狀態下,克拉霉素吸收完全,由于膽汁再攝取增強,73.7%劑量比例吸收分布在十二指腸,22.1%分布在空腸。
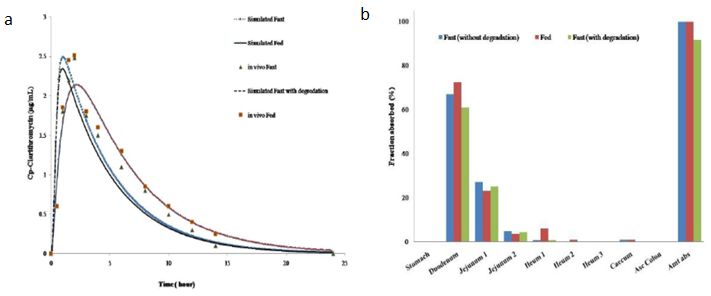
圖3 a:空腹和餐后試驗下克拉霉素模擬和觀測血漿濃度;b:空腹和餐后試驗下克拉霉素的吸收分布
? 敏感性分析:選擇參數溶解性、滲透性、胃排空速率、胃腸pH、胃排空時間、總清除率、肝腸首過效應(FPE)對空腹及餐后試驗條件下克拉霉素的吸收進行敏感性分析。根據結果,清除率、肝臟首過效應和腸道首過效應對克拉霉素的Cmax及AUC具有顯著影響。然而,溶解性、滲透性、胃腸pH對克拉霉素的吸收無影響。餐后試驗條件下,Cmax受胃排空速率影響,Cmax隨胃滯留時間延長而降低。經敏感性分析發現,廣泛的肝臟代謝和腸道首過效應可能會導致PK參數大幅度的下降,給藥后肝血流量增加也會降低肝提取率并增大藥物生物利用度。
02、虛擬BE模擬
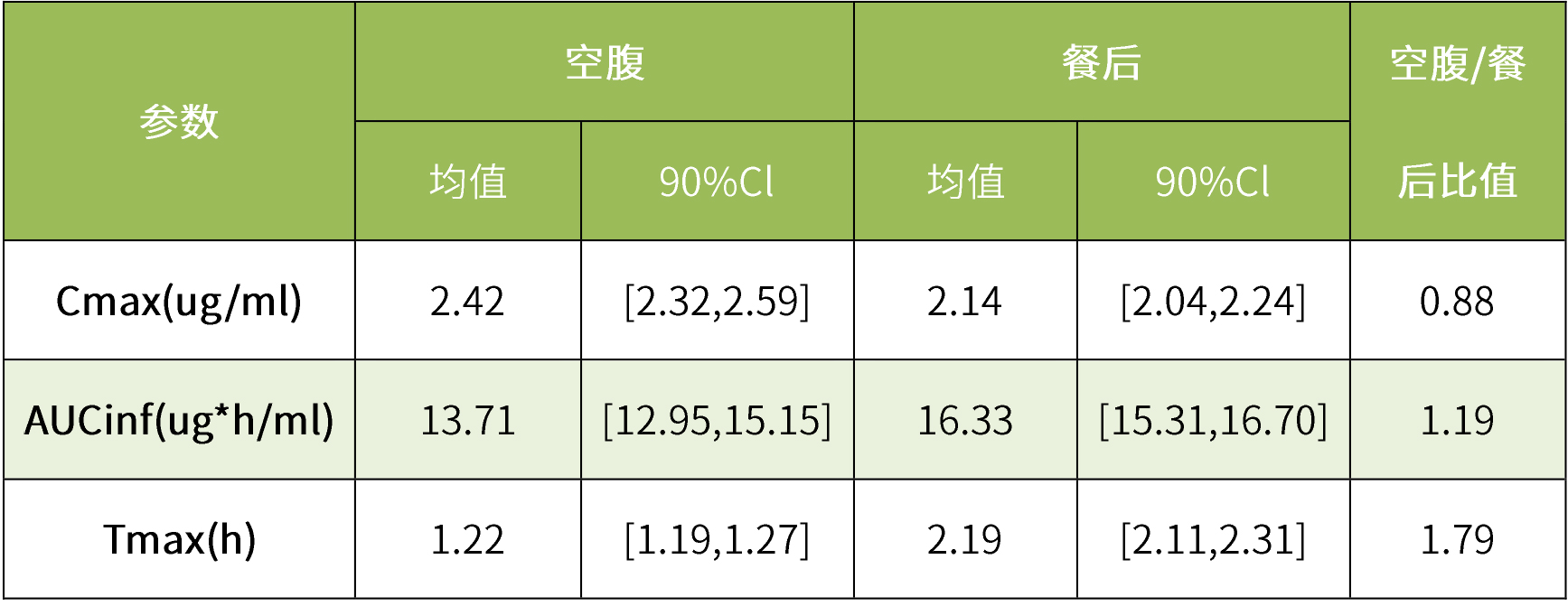
虛擬BE模擬結果表明,食物對克拉霉素生物利用度無顯著影響。克拉霉素片在空腹和餐后狀態下的平均血漿濃度-時間曲線均落在90%置信區間內。虛擬人群試驗表明,食物不會顯著改變克拉霉素片的Cmax和AUC,空腹與餐后的Cmax和AUC的90%置信區間均在生物等效性限值內。
表4 克拉霉素的虛擬食物影響研究
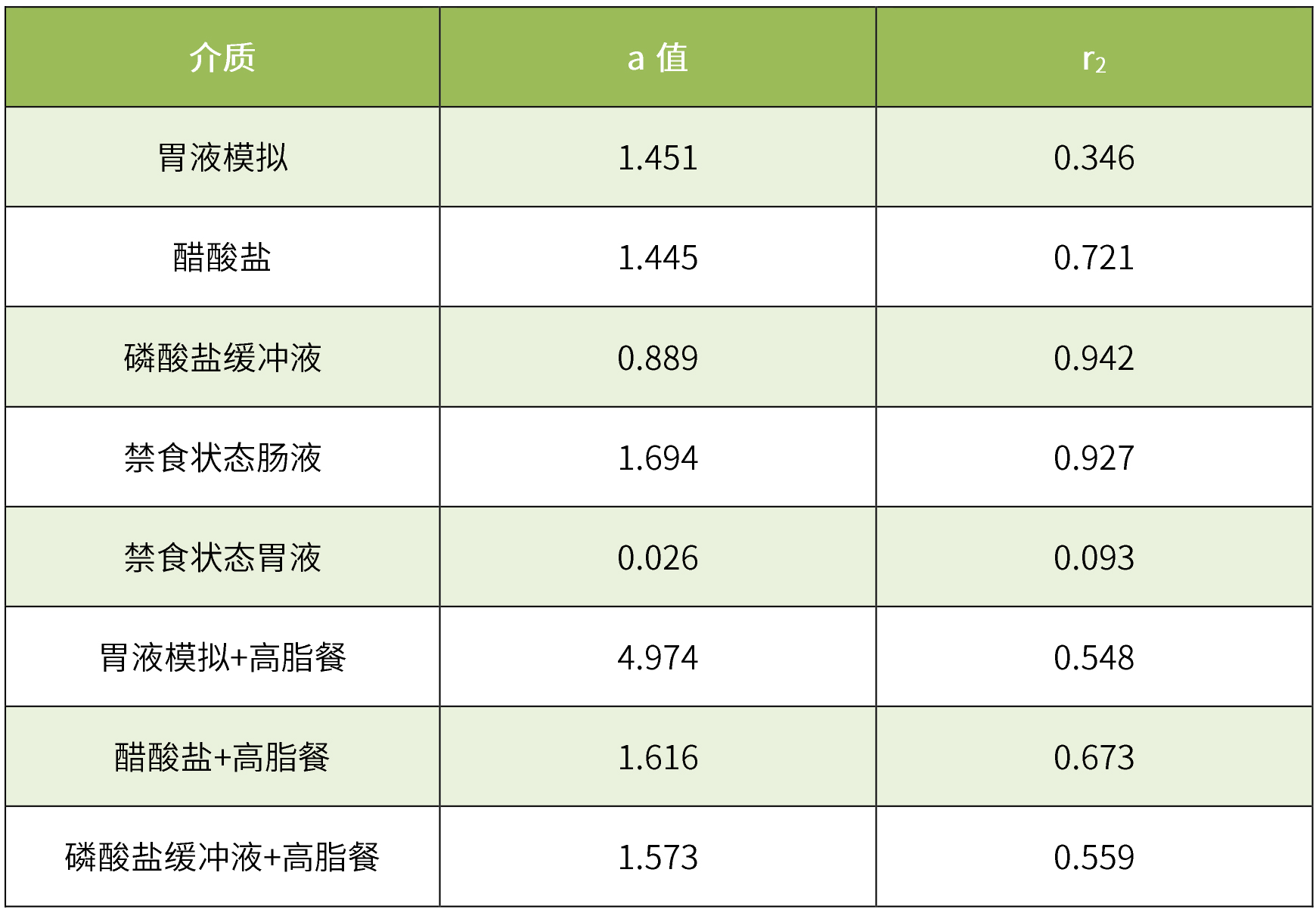
采用去卷積方法,利用機械吸收模型,將空腹和餐后狀態下的體外溶出數據和相應的體內血漿濃度關聯起來,建立體內外相關性。在磷酸鹽緩沖液、醋酸鹽和禁食狀態腸液介質中成功建立了IVIVR關系,相關系數(r2)值接近于1。但是,在HCl介質中體內外相關性較差(r2=0.3),餐后狀態下不同介質的體外溶出與體內相關性均不佳。
表5 IVIVR統計參數
模型驗證
我司擁有國內首個體內外橋接(IVIVR)研發平臺,不僅擁有Gastroplus軟件,而且完成了克拉霉素片仿制藥的申報。小編這里運用GastroPlus軟件的PBPK模型對克拉霉素片進行模型驗證。將我司獲得的餐后狀態下的體內PK數據輸入該模型中,模擬結果見表6。根據結果可知,我司取得的餐后PK數據與模型觀測值擬合度極高,誤差小于10%,二者血漿濃度藥時曲線也幾乎完全吻合,標明該模型具有良好的預測性。
表6 研發中克拉霉素片與模型擬合結果
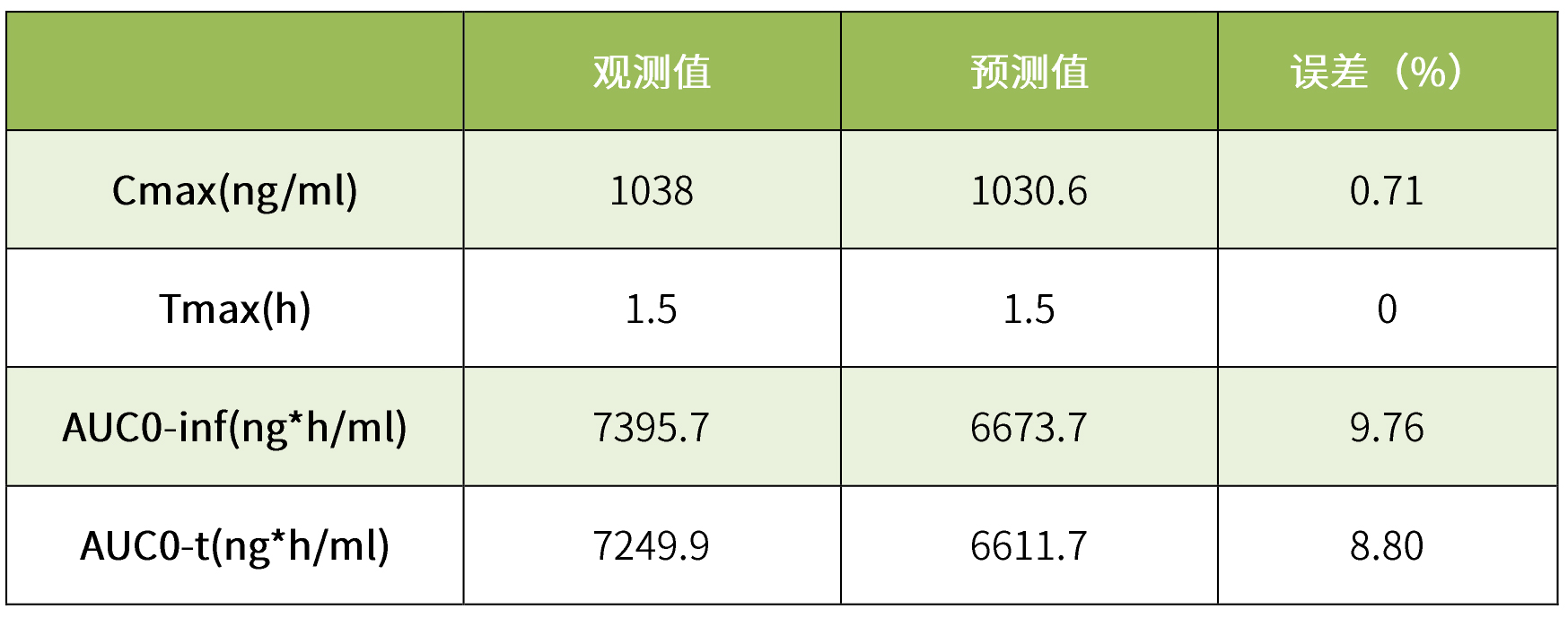
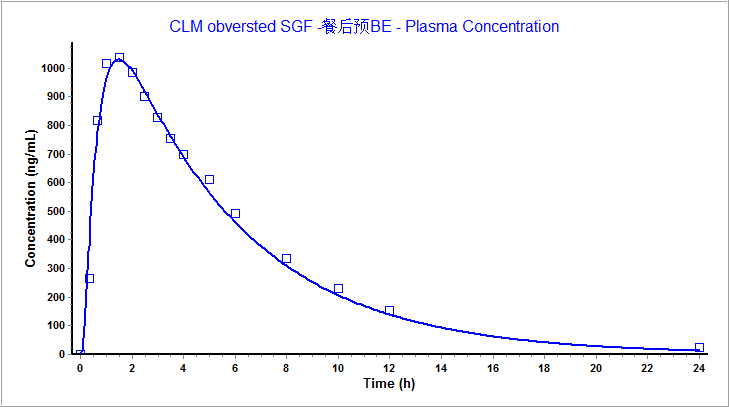
圖5 克拉霉素片在餐后狀態下與模型血漿濃度藥時曲線擬合圖
四、 討論
本研究采用GastroPlus建模和體外試驗,研究了食物對克拉霉素口服生物利用度的影響機制,pH值、膽酸鹽和食物對克拉霉素的口服吸收均具有顯著影響。
-
酸性條件下,克拉霉素與鹽酸反應形成凝膠,使其崩解時間變長。該凝膠層在中性介質中溶解,可加快藥物崩解,促進藥物釋放。
-
在生理相關介質內,膽酸鹽存在時克拉霉素的釋放較常規介質中低很多,是因為大環內酯類抗生素與膽汁鹽發生絡合作用,形成的絡合物會減少藥物釋放,但膽汁的再攝取會增加進入血液的克拉霉素比例。
-
研究表明食物對藥物的釋放和片劑的崩解具有顯著影響,由于pH和膽酸鹽濃度變化,含有標準高脂餐的介質和生理相關介質中的溶出差異顯著。
空腹條件下,短暫的胃滯留時間和較長的片劑崩解時間減少了藥物的釋放,但在進入腸腔后,克拉霉素穩定性改善、片劑崩解快速以及藥物溶出均會促進藥物的吸收。另一方面,食物攝入后胃內pH升高,克拉霉素穩定性增強,清除率降低,胃滯留時間延長,這些也會延長藥物的溶解時間。與空腹相比,進食會降低肝腸首過效應和克拉霉素總清除率,這有助于增大藥物的口服生物利用度。然而,小腸中膽汁鹽濃度的增加會減慢克拉霉素的溶解速率,使得克拉霉素在餐后條件下不完全吸收。從生理學角度看,克拉霉素在磷酸鹽緩沖液、醋酸鹽和禁食狀態腸液介質中均具有較強的體內外相關性。
GastroPlus軟件模擬的濃度-時間曲線與體內觀測數據吻合良好,該模型具有較好的預測性。小編運用實際PK數據進行模型驗證,也表明該模型預測性良好。這些發現表明,體外和PBPK方法可作為預測口服藥物食物效應的可靠工具,在開發仿制藥過程中,可無需過多的耗時和進行昂貴的臨床研究。
-END-


轉載聲明:未經本網或本網權利人授權,不得轉載、摘編或利用其他方式使用上述作品。已經本網或本網權利人授權使用作品的,應在授權范圍內使用,并注明“來源:新領先醫藥科技”。

 010-61006450
010-61006450 聯系地址:
聯系地址: 技術市場部:
技術市場部: 010-61006450
010-61006450
