 Hotline服務(wù)熱線:010-61006450
Hotline服務(wù)熱線:010-61006450
 簡體中文
簡體中文一文教你讀懂FDA審評報告!
美國FDA藥品數(shù)據(jù)庫包含1939年起的絕大多數(shù)批準(zhǔn)藥品,1998年之后批準(zhǔn)的藥品全部信息。Drugs@FDA作為FDA網(wǎng)站中一個數(shù)據(jù)庫主要可以幫助我們完成相關(guān)信息的查找。
審評報告詳細(xì)查詢流程:(以阿昔替尼片為例)
- 首先進(jìn)入Drugs@FDA的搜索欄輸入阿昔替尼片的活性成分“axitinib”,然后點(diǎn)擊提
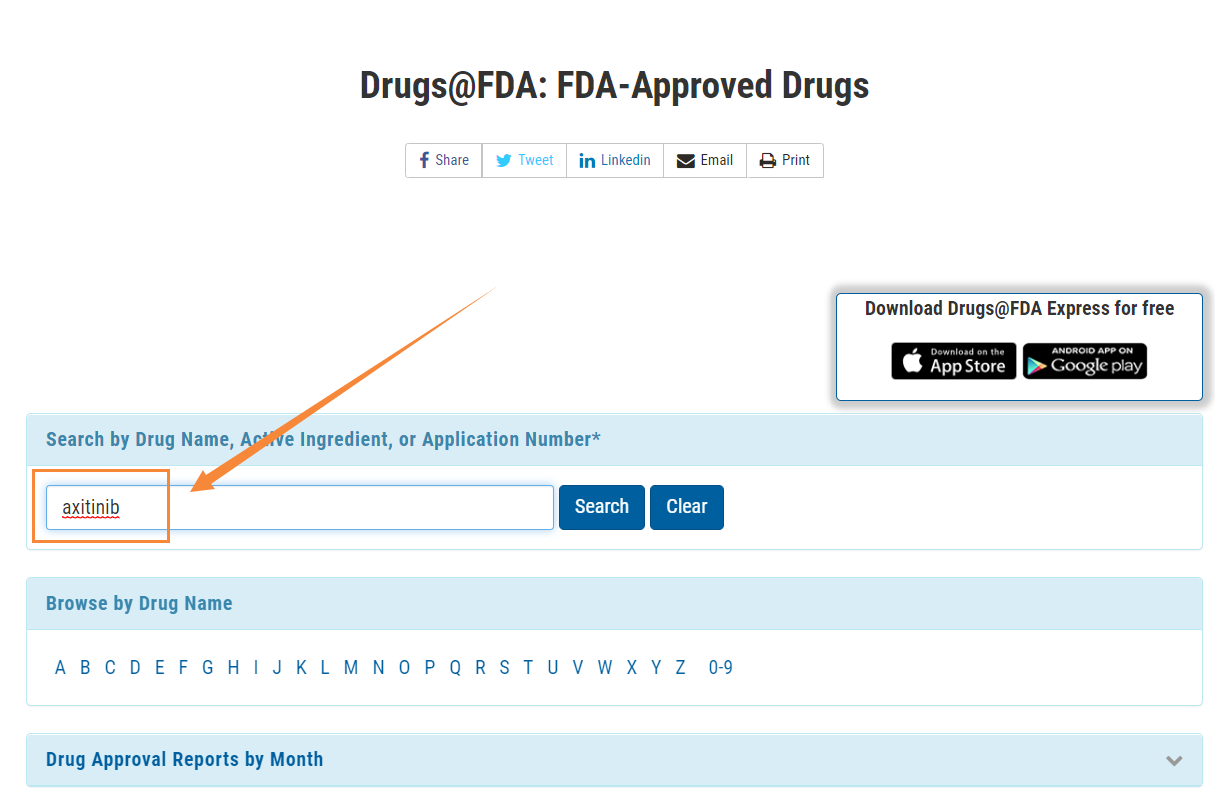
- 進(jìn)入”Drug Details”,看到藥品詳細(xì)信息,點(diǎn)擊”Approval Date(s) and History, Letters, Labels, Reviews for NDA 202324”,在菜單欄目選擇“Review”,即可看到相應(yīng)的審評報告

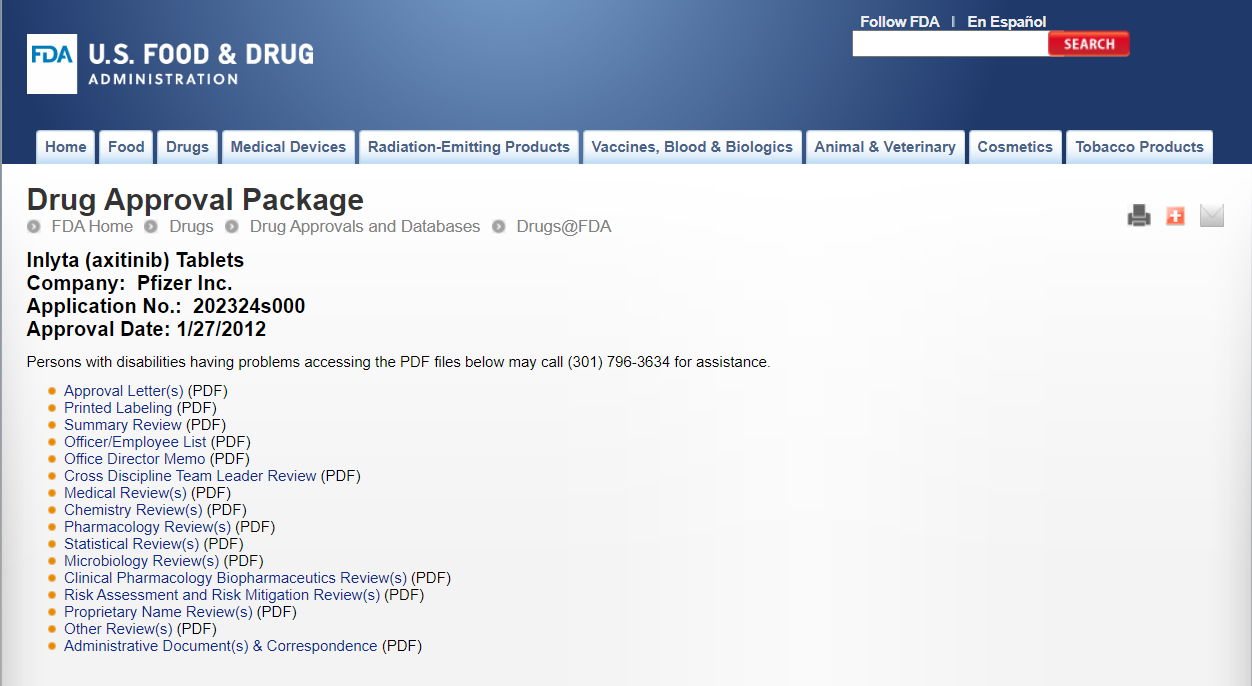
輝瑞公司研發(fā)的阿昔替尼片審評報告
FDA審評報告中一般包含研究內(nèi)容的報告如下:化學(xué)審查報告、藥理學(xué)審查報告、臨床藥理學(xué)/生物藥劑學(xué)審查報告、醫(yī)學(xué)審查報告等。
化學(xué)審查報告是對該藥物原料藥與制劑合成/生產(chǎn)工藝、理化性質(zhì)研究、質(zhì)量控制體系研究、穩(wěn)定性研究等方面的審查結(jié)論。在阿昔替尼片劑的化學(xué)審評報告(Chemistry Review)中,審評員總結(jié)概括的內(nèi)容涉及以下方面:

該部分研究內(nèi)容將貫穿新藥研發(fā)的整個過程,從化合物的發(fā)現(xiàn)、篩選、動物實(shí)驗(yàn),合成路線優(yōu)化及制劑初步開發(fā),臨床前的制劑優(yōu)化,以及臨床后可能進(jìn)行的進(jìn)一步制劑優(yōu)化等。
申請人需在新藥上市申請前提交完整的原料藥及制劑研究資料,需要詳細(xì)闡述其原料藥合成路線及其合理性、制劑制備工藝及其合理性、各項質(zhì)量控制方式及其合理性、穩(wěn)定性研究詳細(xì)數(shù)據(jù)及結(jié)論等。
通過原研藥品的Chemistry Review,我們可以直接獲取參比制劑的處方組成及制備工藝,為我們仿制藥的開發(fā)節(jié)約時間、提高效率。不過,很多時候,由于涉及信息保密,原研化學(xué)審評報告中的很多關(guān)鍵信息會被打上陰影。
藥理學(xué)審評報告含以下內(nèi)容:


該部分詳細(xì)記載了新藥臨床研究審批(IND)申請前藥物在動物體內(nèi)進(jìn)行的、能夠證明擬申報藥物在體內(nèi)安全有效的詳細(xì)試驗(yàn)數(shù)據(jù)及結(jié)論,包括藥理學(xué)研究、藥代動力學(xué)研究、安全藥理學(xué)研究、毒理學(xué)研究、藥效學(xué)研究等。該部分內(nèi)容對仿制藥開發(fā)可參考意義不大,不過了解一下該藥物的作用機(jī)制及不良反應(yīng),或許可以更清楚參比制劑劑型、劑量、服用方式、處方工藝等設(shè)計的理由。
完成動物水平的安全有效性評價后,需要向FDA提交IND申請,方可開展臨床試驗(yàn),臨床藥理學(xué)審查報告就是對藥物臨床試驗(yàn)(包括I期、II期、Ⅲ期)的審查結(jié)論。
在阿昔替尼片劑的臨床藥理學(xué)/生物藥劑學(xué)審評報告中,研究人員/審評員羅列出該藥物在臨床階段所進(jìn)行的各項臨床研究的名稱和概要。(至少包含16項1期臨床、4項2期臨床、1項3期臨床研究)
以“問題+回答”的方式,對該部分需要關(guān)注的重點(diǎn)內(nèi)容進(jìn)行了概括,包括:

對于仿制藥開發(fā),臨床藥理學(xué)/生物藥劑學(xué)審評報告也是我們不可忽略的參考資料。仿制藥上市需要與參比制劑實(shí)現(xiàn)體內(nèi)生物等效,而該文件中所包含的原研不同劑量、空腹/餐后、不同劑型等的藥代動力學(xué)數(shù)據(jù)將有助判斷該藥物體內(nèi)生物等效的風(fēng)險,指導(dǎo)制劑開發(fā)及BE試驗(yàn)方案設(shè)計。
阿昔替尼片劑的醫(yī)學(xué)審評報告中,詳細(xì)列出了3期臨床試驗(yàn)的方案設(shè)計、合格標(biāo)準(zhǔn)、入組人員信息、療效性數(shù)據(jù)及分析(無進(jìn)展生存期分析、中位生存期分析、總生存期分析、客觀緩解率、緩解持續(xù)時間)、安全性數(shù)據(jù)及分析(暴露量、死亡率、由于不良事件而停藥事件、顯著的不良事件、安全性總結(jié))及其他臨床試驗(yàn)期間對人體各項機(jī)能的檢測數(shù)據(jù)等。
醫(yī)學(xué)審查報告也是對臨床試驗(yàn)的審查報告,但與臨床藥理學(xué)/生物藥劑學(xué)審查報告不同的是,醫(yī)學(xué)審查報告重點(diǎn)關(guān)注并詳細(xì)分析關(guān)鍵性3期臨床所反映出的該藥物療效性和安全性信息。
該部分內(nèi)容新藥研發(fā)時可參考用來了解FDA審評員的關(guān)注點(diǎn)、優(yōu)化自己的方案設(shè)計。
結(jié)語
FDA新藥審評報告是醫(yī)藥研發(fā)領(lǐng)域重要的信息來源,新藥研發(fā)可以參考相似藥物的審評報告進(jìn)行實(shí)驗(yàn)設(shè)計和數(shù)據(jù)把控,仿制藥開發(fā)則可以利用參比制劑的審評報告獲取關(guān)鍵信息來提高仿制藥開發(fā)的成功率、節(jié)約開發(fā)周期。
-END-


轉(zhuǎn)載聲明:未經(jīng)本網(wǎng)或本網(wǎng)權(quán)利人授權(quán),不得轉(zhuǎn)載、摘編或利用其他方式使用上述作品。已經(jīng)本網(wǎng)或本網(wǎng)權(quán)利人授權(quán)使用作品的,應(yīng)在授權(quán)范圍內(nèi)使用,并注明“來源:新領(lǐng)先醫(yī)藥科技”。

 010-61006450
010-61006450 聯(lián)系地址:
聯(lián)系地址: 技術(shù)市場部:
技術(shù)市場部: 010-61006450
010-61006450
