 Hotline服務(wù)熱線:010-61006450
Hotline服務(wù)熱線:010-61006450
 簡體中文
簡體中文收藏!IND注冊流程及資料要求(附詳細(xì)流程圖)
據(jù)藥智數(shù)據(jù)梳理,2021年新藥注冊申請共1933件,同比2020年增長68.09%;其中申請臨床1756件,申請上市177件。且值得提及的2021年,國產(chǎn)新藥首次IND品種數(shù)量超過600個;首次獲批的國產(chǎn)新藥數(shù)量達到了23個。中國正在向著國際創(chuàng)新藥研發(fā)的中堅力量發(fā)展。
根據(jù)美國國立衛(wèi)生研究院的數(shù)據(jù)顯示,我國已經(jīng)是世界上開展臨床試驗數(shù)量第二多的國家。然而,新藥研發(fā)從藥物發(fā)現(xiàn)到申報上市是一個充滿冒險與挑戰(zhàn)且周期漫長的過程;其中IND申請是新藥研發(fā)生命周期至關(guān)重要的一環(huán)。
那么該如何提高IND申報通過率?什么情況下需要提交 IND 申請,又需要提前準(zhǔn)備些什么資料?是創(chuàng)新藥企加快新藥進入臨床研究階段仍亟需思考與探討的問題。今日,筆者就帶領(lǐng)大家一起學(xué)習(xí)IND注冊流程及資料要求,助力藥物研發(fā)。
IND(Investigational new drug),一般是指尚未經(jīng)過上市審批,正在進行各階段臨床試驗的新藥。IND申請,即新藥研究申請,目的在于向藥監(jiān)部門提供數(shù)據(jù)證明藥物具備開展臨床試驗的安全性和合理性,獲準(zhǔn)后方可開展臨床試驗。
IND注冊流程
新藥研究上市主流程
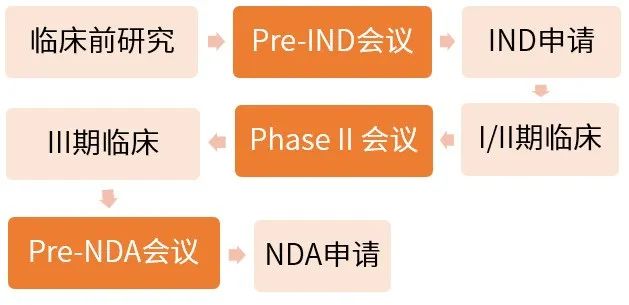
Pre-IND會議
根據(jù)國家藥監(jiān)局藥審中心發(fā)布的關(guān)于《藥物研發(fā)與技術(shù)審評溝通交流管理辦法》的通告(2020年第48號)及國家藥品監(jiān)督管理局發(fā)布的《關(guān)于調(diào)整藥物臨床試驗審評審批程序的公告》(2018年第50號),溝通交流會在藥品上市過程中具有重要意義。
溝通交流,系指在藥物研發(fā)過程中,經(jīng)申請人提出,由藥審中心項目管理人員與申請人指定的藥品注冊專員共同商議,并經(jīng)藥審中心適應(yīng)癥團隊同意,就現(xiàn)行藥物研發(fā)與評價指南不能涵蓋的關(guān)鍵技術(shù)等問題所進行的溝通交流。適用于創(chuàng)新藥物、改良型新藥、生物類似藥、復(fù)雜仿制藥以及一致性評價品種等研發(fā)過程和注冊申請中的溝通交流。會議最終形成的共識可作為研發(fā)和評價的重要依據(jù)。
01溝通交流會會議分類:
溝通交流會分為I類、II類、III類三種會議類型,申請人可在臨床研發(fā)不同階段就關(guān)鍵技術(shù)問題提出溝通交流申請,每類會議針對不同情況開展,其中Pre-IND會議屬于II類會議。
I類:
-
藥物臨床試驗過程中遇到的重大安全性問題;
-
突破性治療藥物研發(fā)過程中的重大技術(shù)問題;
-
其他規(guī)定情形。
II類:
-
新藥臨床試驗申請前會議;
-
藥物Ⅱ期臨床試驗結(jié)束/Ⅲ期臨床試驗啟動前會議;
-
新藥上市許可申請前會議;
-
風(fēng)險評估和控制會議。
※ 特別注意:對于技術(shù)指南明確、藥物臨床試驗有成熟研究經(jīng)驗,申請人能夠保障申報資料質(zhì)量的,或國際同步研發(fā)的國際多中心臨床試驗申請,在監(jiān)管體系完善的國家和地區(qū)已經(jīng)獲準(zhǔn)實施臨床試驗的,申請人可不經(jīng)溝通交流直接提出臨床試驗申請。
III類:
除Ⅰ類和Ⅱ類會議之外的其他會議。
02申請溝通交流會會議流程:
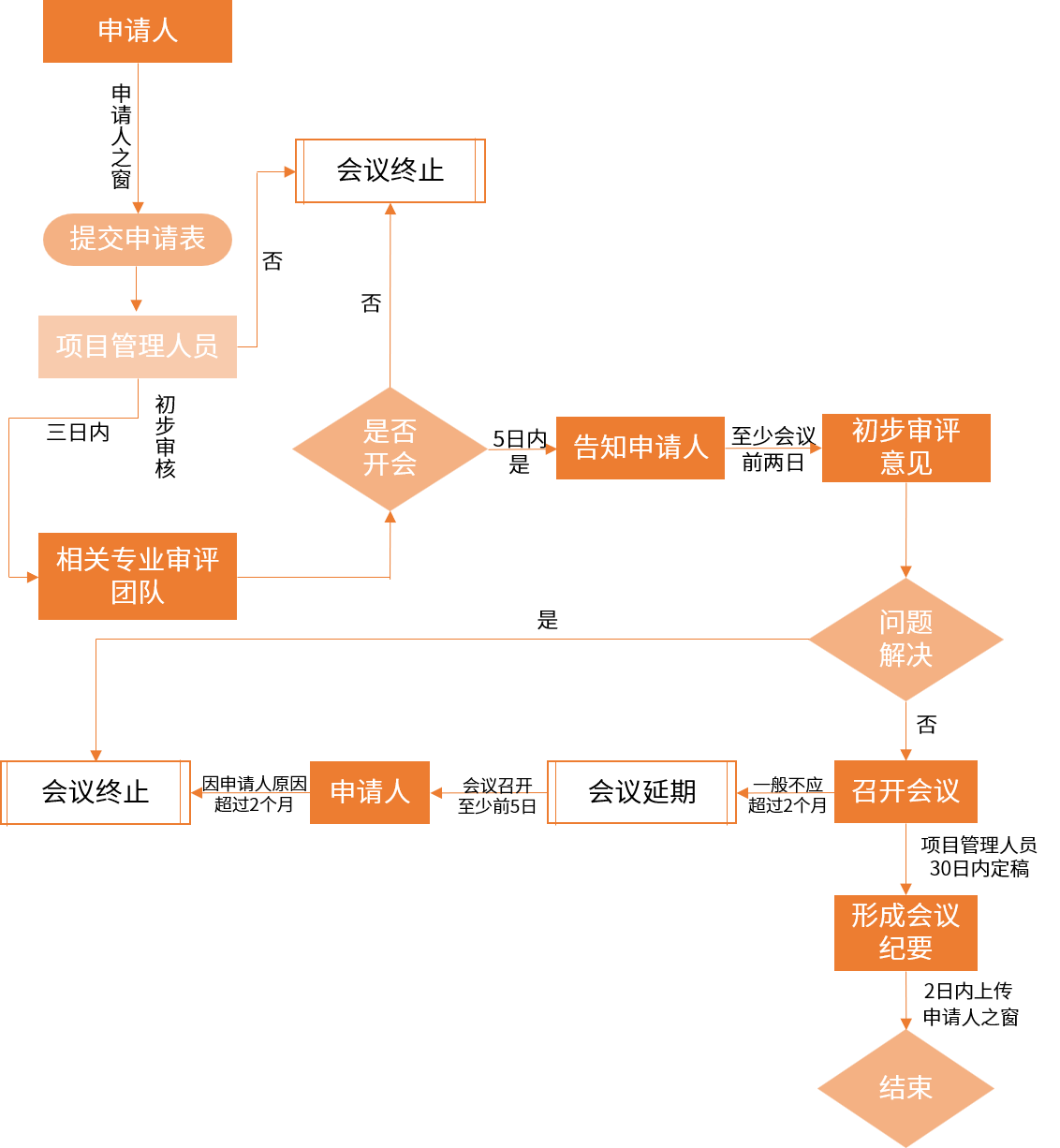
※ 注:從申請人申請到召開會議,I類會議需要在30日內(nèi),II類會議需60日內(nèi),III類會議需75日內(nèi)開展。
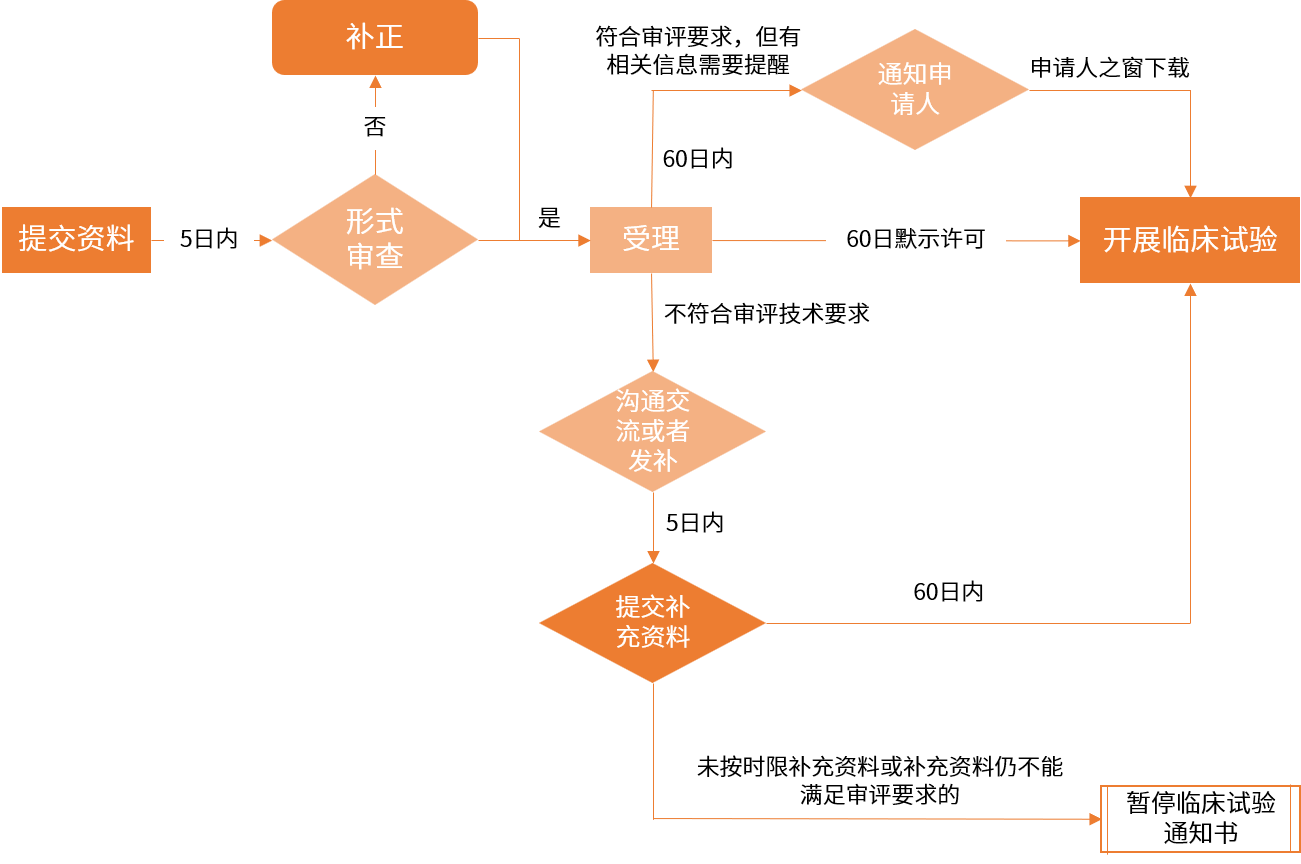
IND受理與審評審批流程
IND注冊資料要求
根據(jù)國家藥品監(jiān)督管理局《關(guān)于調(diào)整藥物臨床試驗審評審批程序的公告》(2018年第50號)及原食品藥品監(jiān)管總局發(fā)布的關(guān)于《新藥I期臨床試驗申請技術(shù)指南》的通告(2018年第16號),IND申請需準(zhǔn)備的資料包括以下:

介紹性說明和總體研究計劃
01介紹性說明:
包括新藥的名稱、所有的活性成分、藥理作用類別、結(jié)構(gòu)式(如果已知)、劑型、制劑處方、給藥途徑、臨床試驗?zāi)康牡取H绻兴芯克幬镉糜谂R床的經(jīng)驗,應(yīng)提供簡短概述,包括在其他國家的研究和上市的經(jīng)驗;若沒有,標(biāo)題下寫“無”。
02總體研究計劃:
總結(jié)申請臨床試驗方案的設(shè)計依據(jù),主要為擬定的適應(yīng)癥、受試者人群、受試者數(shù)量、給藥方案(劑量、給藥間隔、給藥持續(xù)時間等)、藥物安全性評價方法、風(fēng)險控制計劃等,根據(jù)已有信息預(yù)期的任何安全性(重要的已確定風(fēng)險、重要的潛在風(fēng)險、重要的缺失的資料等)的風(fēng)險論證。
研究者手冊
包括封面頁、目錄、保密聲明、概述、新藥名稱與理化性質(zhì)、非臨床研究結(jié)果(藥理作用、毒理作用、非臨床藥代動力學(xué)研究)、已有臨床研究或使用資料(人體藥物代謝動力學(xué)、有效性、安全性及上市情況)、其他及參考文獻。
臨床試驗方案
包括研究背景,試驗?zāi)康模A(yù)計參加的受試者數(shù)量,入選標(biāo)準(zhǔn)和排除標(biāo)準(zhǔn)描述,給藥計劃描述,檢測指標(biāo)、對受試者安全性評價至關(guān)重要的相關(guān)試驗詳細(xì)信息,中止研究的毒性判定原則及試驗暫停標(biāo)準(zhǔn)等。
藥學(xué)研究信息
重點關(guān)注對計劃研究的受試者安全性相關(guān)的藥學(xué)研究信息,出現(xiàn)以下藥學(xué)問題應(yīng)暫緩臨床試驗:
-
新藥化學(xué)結(jié)構(gòu)或制劑輔料具有已知毒性或極可能具有毒性;
-
在計劃實施的整個I期臨床試驗項目期間,新藥不能保持穩(wěn)定性;
-
新藥的雜質(zhì)特征顯示具有潛在毒性,或者新藥中含量在鑒定限以上的雜質(zhì)未進行充分鑒定且未對其潛在的毒性進行評估;
-
存在動物源性成分的生物安全性問題;
-
主細(xì)胞庫或工作細(xì)胞庫未經(jīng)過充分鑒定。
非臨床研究信息
-
非臨床研究綜述:已完成的非臨床研究的概要信息,各項試驗可采用列表形式列出;
-
藥理學(xué)研究的總結(jié):體內(nèi)外藥理作用及其作用機制、次要藥效學(xué)信息、藥效與暴露關(guān)系的研究信息;
-
毒理學(xué)研究的總結(jié):毒性反應(yīng)的程度、嚴(yán)重性和持續(xù)時間,劑量相關(guān)性、可逆性,種屬及性別差異,特別關(guān)注重復(fù)給藥毒性反應(yīng)信息、動物死亡、病理學(xué)檢查、局部耐受性、其他需特別說明問題;
-
藥代動力學(xué)的總結(jié):分析方法的可行性,藥代動力學(xué)/毒代動力學(xué)參數(shù),吸收與組織分布、代謝、排泄,藥效和毒性問題引起的生理變化,如疾病狀態(tài)的影響、抗體生成、交叉反應(yīng)性等;
-
各項研究報告:提供已經(jīng)獲得的藥理作用、毒理研究和藥代動力學(xué)的各項研究報告;
-
其他。
既往臨床使用經(jīng)驗說明
-
如果有既往的臨床使用經(jīng)驗,申請人應(yīng)提供相關(guān)信息概述;
-
如果研究藥物曾經(jīng)在中國或者其他國家開展了臨床研究或者已經(jīng)上市,應(yīng)提供與擬開展試驗的安全性或者擬開展試驗依據(jù)有關(guān)的詳細(xì)信息;
-
應(yīng)提供與擬開展試驗的安全性有關(guān)的所有已發(fā)表文獻資料或者對研究藥物擬開展適應(yīng)癥研究的有效性評價數(shù)據(jù),包括與研究藥物既往臨床使用經(jīng)驗有關(guān)的參考文獻列表或者重要的支持性文獻。除此之外,還應(yīng)根據(jù)已有信息綜合評估擬開展的臨床研究,這將有助于支持臨床研究的劑量、用藥持續(xù)時間、藥物組合、受試人群的選擇。
-END-



轉(zhuǎn)載聲明:未經(jīng)本網(wǎng)或本網(wǎng)權(quán)利人授權(quán),不得轉(zhuǎn)載、摘編或利用其他方式使用上述作品。已經(jīng)本網(wǎng)或本網(wǎng)權(quán)利人授權(quán)使用作品的,應(yīng)在授權(quán)范圍內(nèi)使用,并注明“來源:新領(lǐng)先醫(yī)藥科技”。

 010-61006450
010-61006450 聯(lián)系地址:
聯(lián)系地址: 技術(shù)市場部:
技術(shù)市場部: 010-61006450
010-61006450
