 Hotline服務(wù)熱線:010-61006450
Hotline服務(wù)熱線:010-61006450
 簡體中文
簡體中文深度:一文看懂中美I類新藥審評差異
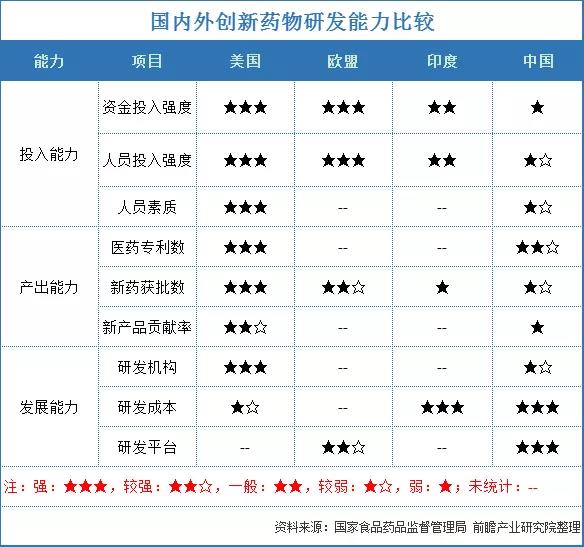
創(chuàng)新藥的數(shù)量和質(zhì)量代表一個國家的創(chuàng)新能力,也是國家經(jīng)濟的增長點。目前我國已成為世界第一大原料藥生產(chǎn)和出口國、第二大非處方藥市場及第三大醫(yī)藥市場,但我國的新藥研發(fā)與歐美國家相比相對薄弱。2020年,美國CDER總共批準53個新藥,歐盟EMA共批準42個新藥,中國NMPA共批準48個新藥上市,其中國產(chǎn)新藥20個(比2019年增加6個),進口新藥28個。
與發(fā)達國家相比,在許多指標上我國創(chuàng)新藥物研發(fā)仍處于明顯劣勢。無論是研發(fā)投入能力還是產(chǎn)出方面與發(fā)達國家均有一定差距。本文通過對比我國與美國I類新藥相關(guān)政策與臨床研究審批等方面的差異,發(fā)現(xiàn)可借鑒的藥物臨床研究審批經(jīng)驗,促進我國新藥研究發(fā)展。
中美新藥定義的差異
藥品,是指用于預(yù)防、治療、診斷人的疾病,有目的地調(diào)節(jié)人的生理機能并規(guī)定有適應(yīng)癥或者功能主治、用法和用量的物質(zhì)。
由于經(jīng)濟發(fā)展水平不同,中美兩國對新藥的定義也略有不同。
美國新藥定義
美國食品藥品監(jiān)督管理局(Food and Drug Administration)[1]對新藥的定義是“凡在1938年的《食品、藥品和化妝品法》(Federal Food, Drug, and Cosmetic Act)公布后提出的任何具有化學(xué)組分的藥品,其說明書中提出的用途未被經(jīng)培訓(xùn)并有評價經(jīng)驗的專家普遍承認其安全性和有效性的;或雖其安全性和有效性已被普遍承認,但尚未在大范圍或長時間使用的,稱為“新藥”。
中國新藥定義
2015年8月以前我國對新藥的審批按照《藥品注冊管理辦法》[2]執(zhí)行,其中對新藥明確闡述為“未曾在中國境內(nèi)上市銷售的藥品的注冊申請”;“對已上市藥品改變劑型、改變給藥途徑、增加新適應(yīng)證的藥品注冊按照新藥申請的程序申報”;“生物制品按照新藥申請的程序申報”。
通過對比可見,我國新藥申請范疇相對較寬,新藥申請程序不僅包括創(chuàng)新藥物的申請,還包括按新藥申請程序的仿制藥物的申請。2015年8月18日國務(wù)院頒布了《關(guān)于改革藥品醫(yī)療器械審評審批制度的意見》(國發(fā)〔2015〕44號,以下簡稱《意見》),其中將新藥明確定義為“未在中國境內(nèi)外上市銷售的藥品”[3]。并將新藥分為創(chuàng)新藥和改良型新藥。此改變反映了我國在新藥審評制度方面逐漸與國際接軌,將有力促進我國創(chuàng)新藥物的研制。

中美新藥審評的差異
我國醫(yī)藥產(chǎn)業(yè)發(fā)展較快,藥品質(zhì)量和標準不斷提高,公眾用藥需求得到較好的滿足。但同時,藥品在審評過程中存在的問題也越來越凸顯。中美兩國對新藥審評的不同主要體現(xiàn)在以下幾個方面:
審評程序和申報機制的差異
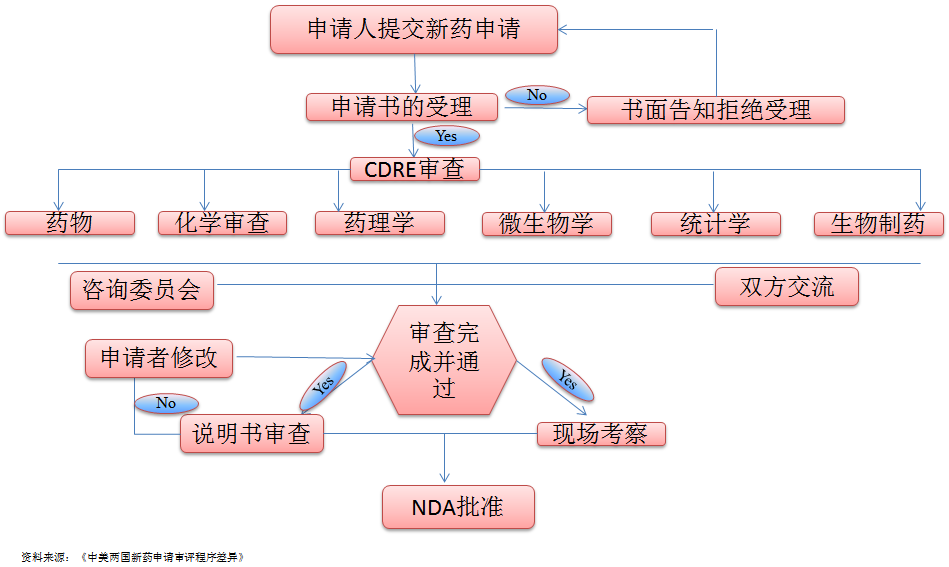
FDA采取直接申報、一級審評的申報機制。美國藥物審評研究中心對新藥申請(New Drug Application,NDA)的申報資料要求[4],首先進行60天的初審(其中包括形式審查),對于形式審查不合格或技術(shù)內(nèi)容存在明顯缺陷的申報資料一律不受理而退審,通過初審后的NDA直接進入復(fù)審(其中包括技術(shù)審查)。
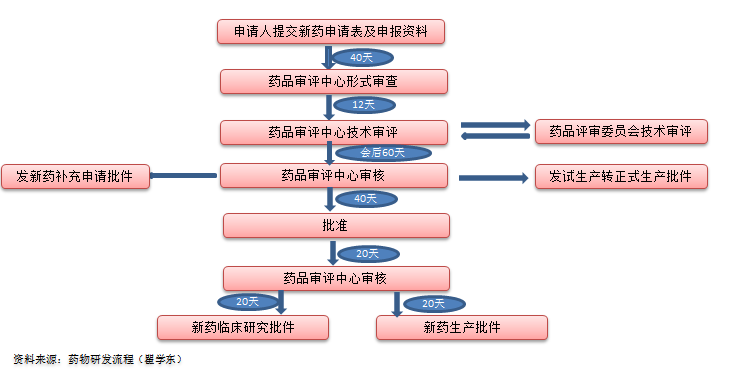
在我國負責(zé)新藥評審的管理機構(gòu)是國家藥品監(jiān)督管理局藥品審評中心(CENTER FOR DRUG EVALUATION,NMPA CDE)。我國采用的是多次申報、二級審評的審評機制。藥品監(jiān)督管理部門負責(zé)形式審查和初審,“受理辦”進行再次形式審查。我國的初審和終審(國家級)不是同一批審評人員(FDA初審和終審工作均為同一批審評人員),這樣在審評工作中的審評規(guī)則不盡相同,而且兩次初審必然會產(chǎn)生審評周期長、審評效率低、審評成本高等一系列問題。如何適應(yīng)我國的國情,改變現(xiàn)有的審評程序、申報機制將是一項艱巨的任務(wù)。
美國新藥申報
中國新藥申報
審評原則的差異
FDA對于申請進行臨床研究(investigational newdrug,IND)的藥品技術(shù)要求比較寬松,只要有初步的質(zhì)量、藥效和動物藥代動力學(xué)研究資料,能夠簡單說明無嚴重的安全性問題,就可批準進行臨床研究。美國新藥申請的退審階段主要集中在I期和II期臨床研究,尤其是在II期臨床研究之后,新藥的淘汰率較高。
我國恰好相反,我國新藥退審主要集中在申請臨床試驗階段[5]。臨床前研究的實驗對象是動物,從動物身上獲得的試驗數(shù)據(jù)雖然對藥物的評價有一定的參考作用,但真正評價一個藥物是否安全有效的主要依據(jù)應(yīng)是從臨床試驗中獲得的數(shù)據(jù),臨床研究才是評價新藥安全有效的關(guān)鍵階段,是決定藥物能否批準上市的試金石。
再評價機制的差異
藥物再評價是對市場藥物進行安全性或有效性評價的研究,是評價藥物安全有效性的一個重要組成部分,也是NDA申請審評的一個重要方面。藥物是否真的安全有效,只有在藥物上市后,通過大范圍的人群長時間使用來驗證。
美國對于上市新藥有非常健全的再評價機制[6],其工作內(nèi)容包括藥品不良反應(yīng)報告和監(jiān)測制度、定期報告制度、上市后臨床試驗及研究制度三部分,在再評價體系下,F(xiàn)DA對上市后藥品的安全風(fēng)險進行實時監(jiān)測,以期有效控制風(fēng)險。
2015年9月10日,國家食品藥品監(jiān)督管理總局發(fā)布的《藥品不良反應(yīng)報告和監(jiān)測檢查指南(試行)》。2018年,我國“藥品上市許可持有人藥品不良反應(yīng)直接報告系統(tǒng)”正式出臺,藥品生產(chǎn)企業(yè)將通過該系統(tǒng)上報不良反應(yīng)。2020年我國根據(jù)《中華人民共和國藥品管理法》及《中華人民共和國疫苗管理法》,制定《藥物警戒質(zhì)量管理規(guī)范(征求意見稿)》,進一步規(guī)范了藥品全生命周期藥物警戒活動[7]。這為掌握新藥上市后的用藥安全提供了很好的依據(jù)。我國已逐漸向發(fā)達國家靠攏。
中美新藥研究申請程序異同
相同點:
中美兩國新藥研究申請程序相同點有:申請新藥注冊都必須進行臨床試驗申請和倫理委員會的審查,對不符合要求的申請都可能導(dǎo)致程序的終止。
差異:
美國的新藥臨床試驗申請評審[8]主要包括兩個階段:第一階段是對申報資料進行形式審查,第二階段是對藥品安全性和有效性進行實質(zhì)性審查和技術(shù)性評價,兩個階段皆由美國FDA下設(shè)的藥品評價和研究中心(CDER)審查。
FDA的評審效率相對更高,我國新藥臨床試驗的技術(shù)評審時限為90個工作日,美國為30個工作日。我國新藥臨床試驗的許可程序包括形式審查、現(xiàn)場核查、初步審查和技術(shù)評審等環(huán)節(jié);美國審核程序簡單,CDER將IND審評重點放在安全性上,F(xiàn)DA收到IND申請自30日內(nèi)未作出暫停臨床研究的決定,即視為默認同意,申請人即可開展臨床研究。
FDA認為IND申請資料有微小缺陷,申請人甚至在對缺陷進行整改的同時進行臨床試驗。暫停臨床研究超過1年或更長時間,并不必然導(dǎo)致新藥臨床試驗申請的失效,可以IND轉(zhuǎn)入“靜止狀態(tài)”。只有當“靜止狀態(tài)”超過5年以上才可能導(dǎo)致終止。由此可見,美國臨床審評的過程是一個動態(tài)過程,即對臨床試驗過程中遇到的安全性問題(包括沒有在預(yù)想范圍內(nèi)的),臨床審評員要不斷做出判斷并反饋給申辦者。
FDA對藥物臨床試驗申請審查不是很嚴,申報的機會更多、門檻更低、行政許可通過率更高。在研究過程中,F(xiàn)DA可以隨時發(fā)現(xiàn)與研究發(fā)起人進行有關(guān)IND不足之處或FDA需要更多數(shù)據(jù)信息的口頭或書面交流,這種交流活動一般視為“建議”,無需要更改計劃或正在進行的臨床研究項目,發(fā)起人也無需答復(fù)FDA。
我國新藥臨床申請由國家食品藥品監(jiān)督管理總局集中受理。申請者必須經(jīng)國家食品藥品監(jiān)督管理總局批準后,獲得藥物臨床試驗批件后方可開展臨床試驗。申請者需要提交審查綜述、藥學(xué)研究、藥理毒理研究、臨床試驗等有關(guān)資料,同時要求所有材料在I期臨床試驗前提交,且在4個月內(nèi)一次性補齊所有材料。
總體來說,我國對臨床研究申請所需提交的資料要求相對嚴格,這與我國以仿制藥開發(fā)為主的行業(yè)背景有關(guān)。對仿制藥來說,不論是藥學(xué)研究資料還是藥理毒理資料均已有前人的研究經(jīng)驗,申報者只要收集、模仿,按要求提交即可。但對新藥(尤其是完全創(chuàng)新藥)來說,臨床試驗還只是探索性的階段,許多研究數(shù)據(jù)(如長期毒性資料)還有待在后期研究中總結(jié)發(fā)現(xiàn)。因此,要求申報者在I期臨床前需提交上述資料,此時耗費大量的時間和精力,從而影響了新藥臨床試驗審批的效率。
中美新藥非臨床研究申請異同
為申請臨床研究而做的非臨床研究中,中國一般包括藥理學(xué)、主要藥效學(xué)和藥代動力學(xué)。而美國對動物研究方面比中國要寬。在毒理學(xué)研究方面,中國規(guī)定做急性毒性和長期毒性試驗,而美國則做急性、亞急性和慢性三種9全身毒性試驗。對于遺傳毒性,中國要求只有當確實需要通過動物長期給藥研究評價人體中藥物暴露所致的潛在致癌性時,才應(yīng)進行致癌試驗[9]。美國則要求,凡致突變試驗陽性或人體使用累計達三個月以上的藥物必須做致癌試驗。

中美新藥臨床研究申請異同
相同點:

在美國,新藥臨床申請試驗分為商業(yè)性和非商業(yè)性兩類。非商業(yè)性申請目的分為研究性、急救性、治療性新藥臨床試驗申請,此類申請高于商業(yè)性申請。而我國新藥臨床研究申請主要目的是商業(yè)性的,即申請新藥通過臨床研究,最終投入生產(chǎn)銷售。

中美新藥優(yōu)惠政策的差異
為了鼓勵新藥研發(fā)和對知識產(chǎn)權(quán)的保護,中美兩國對所研發(fā)的新藥均有優(yōu)惠政策。
美國市場獨占權(quán):
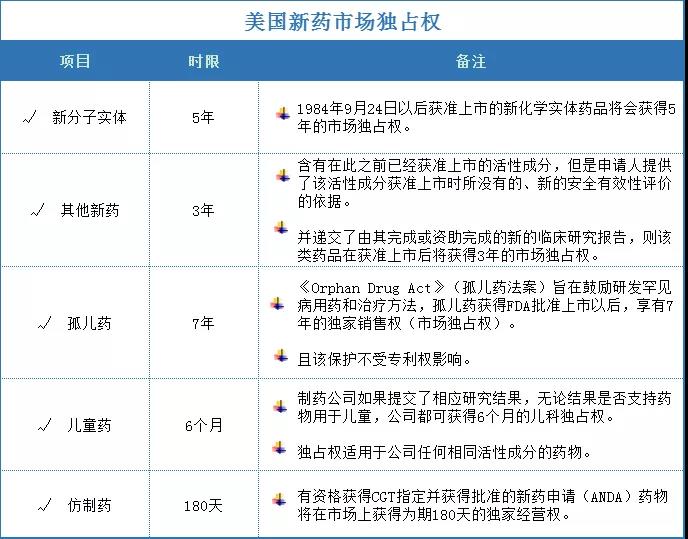
中國:
設(shè)立了對新藥品種3-5年的新藥監(jiān)測期。在新藥監(jiān)測期內(nèi),其他藥品生產(chǎn)企業(yè)不得生產(chǎn)相同品種的藥品。
此外,中美兩國在加速審評審批方面也均有一定的政策鼓勵[10]。
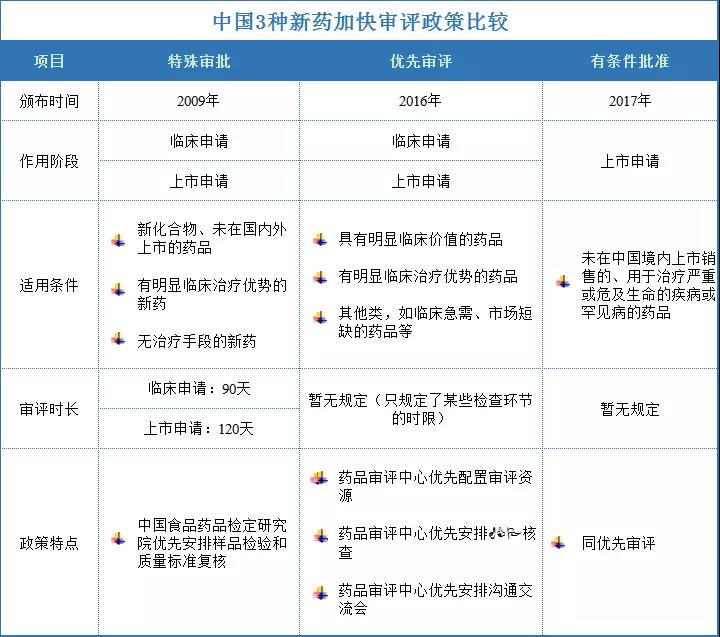
中國新藥上市加快審評審批政策包括4種類型,分別是特別審批、特殊審批、優(yōu)先審評、有條件批準。由于“特別審批”涉及重大突發(fā)衛(wèi)生公共事件,滿足條件的品種很少,因此,通常情況下中國新藥上市加快審評類型可以歸納為3種,分別是特殊審批、優(yōu)先審評、有條件批準。
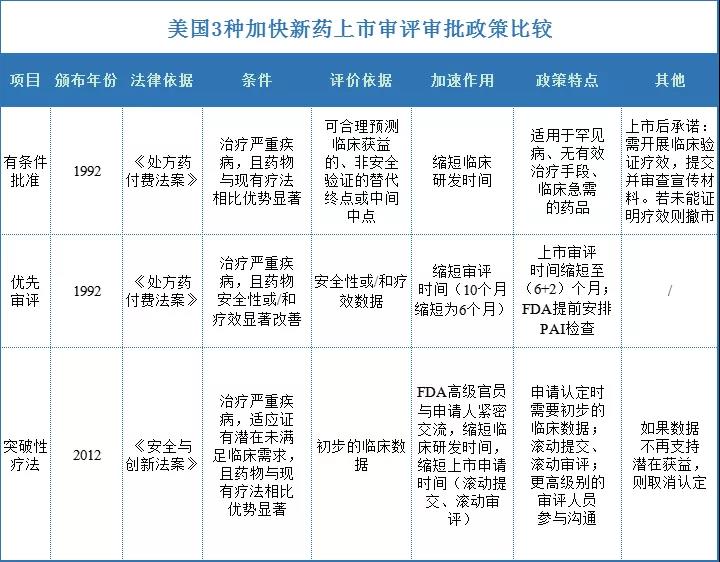
美國的新藥上市加快審評審批的政策有3種,分別是突破性療法(breakthrough therapy)、優(yōu)先審評( priority review)、有條件批準(accelerated approval)。
總結(jié)
2017年以來,我國醫(yī)藥行業(yè)的政策監(jiān)管愈發(fā)嚴格,藥品注冊申請的要求也越來越完善,不管是仿制藥、新藥還是補充申請都要嚴格符合注冊法規(guī)的相關(guān)要求,提供完整、真實且可溯源的全套藥學(xué)研究、非臨床研究和臨床研究資料,促進了國內(nèi)醫(yī)藥行業(yè)發(fā)展從粗放式向精細化的轉(zhuǎn)變,逐步與國際接軌。
提升醫(yī)藥產(chǎn)業(yè)創(chuàng)新能力,鼓勵發(fā)展創(chuàng)新藥物,是國家醫(yī)藥產(chǎn)業(yè)實現(xiàn)跨越式發(fā)展的核心引擎。美國作為世界上新藥研發(fā)能力最強的國家,其新藥研發(fā)的成功經(jīng)驗,對我國具有很大的借鑒意義。學(xué)習(xí)和借鑒發(fā)達國家的成功經(jīng)驗,克服不利因素,以更加積極主動的姿態(tài)投入到創(chuàng)新藥物的研發(fā)中,以實現(xiàn)我國醫(yī)藥產(chǎn)業(yè)跨越式發(fā)展。
參考文獻:
[1]https://www.ecfr.gov/cgi-bin/text-idx?SID=a5e13772d51b5900dec2f52bb91c32e9&mc=true&node=se21.5.310_13&rgn=div8
[2]https://www.nmpa.gov.cn/directory/web/nmpa/xxgk/fgwj/bmgzh/20070710010101571.html
[3]https://www.nmpa.gov.cn/directory/web/nmpa/zhuanti/lshzht/fzhypj/fzhyzhcfg/20150818101201803.html
[4] https://www.fda.gov/medical-devices/premarket-approval-pma/pma-review-process
[5]余菁菁. 中美新藥審批制度的比較研究:一個經(jīng)濟學(xué)的視角.
[6]張琪, 顏建周, 馬旭峰, 等. 美國藥品上市后再評價法律制度實施的研究及其對我國的啟示[J]. 中國藥房, 2019, 30(15): 2017-2022.
[7]《藥物警戒質(zhì)量管理規(guī)范(征求意見稿)》.
[8]https://www.ecfr.gov/cgi-bin/retrieveECFR?gp=&SID=9d8edd80fb9c9d2e7149351a66421212&mc=true&n=sp21.5.312.b&r=SUBPART&ty=HTML#se21.5.312_122
[9] https://www.nmpa.gov.cn/xxgk/fgwj/gzwj/gzwjyp/20100401145801553.html
[10]任曉星, 史錄文. 中美歐新藥上市加快審評審批政策研究[J]. 中國新藥雜志, 2020, 29(9): 9621-971.
-END-
關(guān)于我們:
綜合信息部由20名成員組成,其中總監(jiān)1名、經(jīng)理2名、副經(jīng)理1名、主管1名、藥理專員15名,均來自北京大學(xué)、中國醫(yī)學(xué)科學(xué)院北京協(xié)和醫(yī)學(xué)院等國內(nèi)外知名院校。
綜合信息部主要負責(zé)公司的國際/國內(nèi)項目立項、品種市場信息支持、公司知識產(chǎn)權(quán)維護及撰寫臨床/藥理相關(guān)申報材料,為公司研發(fā)提供信息保障。
部門下設(shè)5個中心,其中仿創(chuàng)藥立項策略中心主要負責(zé)仿制藥、創(chuàng)新藥品種立項;臨床&藥理中心負責(zé)相關(guān)申報材料撰寫;知識產(chǎn)權(quán)中心負責(zé)公司知識產(chǎn)權(quán)維護;大數(shù)據(jù)分析中心負責(zé)國內(nèi)外品種信息分析、企業(yè)產(chǎn)品管線規(guī)劃及戰(zhàn)略合作伙伴個性化信息支持;國際新產(chǎn)品策略中心關(guān)注國際原料藥、裝置、制劑品種信息,提供中美、中歐雙報立項信息。


轉(zhuǎn)載聲明:未經(jīng)本網(wǎng)或本網(wǎng)權(quán)利人授權(quán),不得轉(zhuǎn)載、摘編或利用其他方式使用上述作品。已經(jīng)本網(wǎng)或本網(wǎng)權(quán)利人授權(quán)使用作品的,應(yīng)在授權(quán)范圍內(nèi)使用,并注明“來源:新領(lǐng)先醫(yī)藥科技”。

 010-61006450
010-61006450 聯(lián)系地址:
聯(lián)系地址: 技術(shù)市場部:
技術(shù)市場部: 010-61006450
010-61006450
